Achieving Compliance with 21 CFR Part 11: Managing Electronic Testing Data
When transitioning from paper to electronic record keeping and Part 11, companies often have several solutions to choose from.

All mechanical testing data should be managed in a way to ensure that it follows the ALCOA acronym: the data is attributable, legible, contemporaneous, original, and accurate. Meeting FDA 21 CFR Part 11 is most commonly required for quality control and production laboratories, however it is important for all labs, including R&D, to consider the ALCOA acronym.
FDA 21 CFR Part 11 compliance is mandatory for medical device and pharmaceutical companies that supply products into the United States. Part 11 is focused around electronic data management and electronic signatures. All mechanical testing data should be managed in a way to ensure that it follows the ALCOA acronym: the data is attributable, legible, contemporaneous, original, and accurate.
For medical device and pharmaceutical companies, achieving data compliance is best met through collaboration between the end users and the equipment supplier. The end users know how the equipment needs to fit into their quality management systems (QMS), and the equipment supplier knows how the system functions and how it can technically meet compliance requirements. In order to achieve full compliance there must be a partnership between the technical functionality of the system and the end user’s QMS. This article will summarize the requirements of FDA 21 CFR Part 11 and discusses the challenges and options laboratories face when transitioning from paper records to electronic records retention.
Data archiving is needed so that test results can be reviewed for an audit or if a quality issue arises. Prior to the rise of electronic data storage, companies used paper records to manage their laboratory data. However, as hard drive and cloud storage becomes increasingly affordable, and laboratory equipment specifications increase to provide more data, it has become less and less practical to store test results via paper records. Furthermore, paper records are more susceptible to data integrity issues, as results can be more easily manipulated.
While most companies want to move away from paper records, they are challenged by the task of changing internal processes and writing new standard operating procedures. It can be overwhelming to change procedures that have been in place for decades, especially when laboratory equipment suppliers have different solutions with varying levels of interpretation of what is required by the FDA to meet Part 11.
One of the basic interpretations of FDA 21 CFR Part 11, put in layman’s terms, is to know and prove who does what, when, how, and why. Knowing the ‘who’ is most commonly achieved by requiring users to provide a unique username and password to log in to a software program. By requiring users to log in, the username can be displayed in the audit trail as proof of who is completing an action. For many companies, there is a concern that users could potentially share passwords in order to log into the software as another user. While this type of malicious behavior is rare, one way to help deter it is by using a system that automatically forces users to change their passwords at set intervals. In addition, many software programs have the ability to link username and password criteria to Windows credentials, which is highly beneficial to laboratories since the IT department will then control criteria such as password length, number of characters, and the frequency at which passwords need to be reset. For many companies, giving the IT department control of test system login credential settings helps labs integrate laboratory equipment into their QMS.
After identifying who is using the equipment, laboratories need to know and report on what actions their users are taking. This can be a challenge for laboratories with more complex equipment, as there may be thousands of input fields a user could potentially change. One way to minimize risk is by using a software package that allows labs to customize permissions. Perhaps a system administrator is granted full access to make changes in the software, whereas an operator only has access to run tests. The audit trail for ‘what’ changes the operator makes will therefore be more simplistic and limited to actions like login attempts, entering required data fields such as lot number or batch number, and pressing the ‘start’ and ‘save’ buttons.

Many companies meeting FDA 21 CFR Part 11 require the username along with date and timestamp to be displayed along with results.
The ‘what’ also ties into the ‘how’ a user interfaces with the system. If it takes a user two tries to log into the software, this should be captured in the audit trail. If a user tries to circumvent saving a file in the software but is then forced to save, the end action is that the file is saved, but the fact that the user tried to exit out of the software and was forced to save should also be captured.
The ‘when’ is the most straightforward parameter that should appear in a data audit trail. The ‘when’ is usually a date and timestamp taken from the computer’s clock. This allows for items in the audit trail to be displayed chronologically.
Finally, the ‘why’ is most commonly an electronic note written by the user who is performing the test or making a change to the software. The note is in place to record the user’s reasoning for making a change. This is usually, but not always, accompanied by an electronic sign-off. The ‘why’ helps a lab manager and auditor understand testing records that were captured weeks, months, or even years prior. The ability to understand who does what, when, how, and why, and the ability to have this information available in a readily accessible audit trail, is critical to labs who strive to meet Part 11.
When transitioning from paper to electronic record keeping and Part 11, companies often have several solutions to choose from. Companies can choose to work with the equipment supplier to implement the equipment supplier’s software solution for 21 CFR Part 11, or they can work with a third-party laboratory compliance company that can offer one wrapper solution for all of their equipment. Depending on your lab’s unique needs, one of these options may be more advantageous than the other. However, for complex laboratory equipment, it is most likely best to work with the original equipment supplier for a Part 11 solution. This is because the equipment supplier will know the nuances within their software program and will best be able to guide the quality team in terms of setup and operation. The strength of a third-party solution, which works well for simplistic programs, is that it enables the lab to implement a single software package across the entire laboratory. These third-party solutions are often Windows-based and can be used to lock down directories and prevent malicious tampering with programs. However, one downside of a third-party wrapper solution is that it often lacks the ability to produce a detailed audit trail defining which specific parameters have been changed. These solutions also often require a separate login for electronic sign-offs.
By placing stringent record-keeping requirements on companies, the FDA is seeking to protect consumers, not to overburden medical device and pharmaceutical companies. In fact, for many laboratories which convert from paper records to full electronic records retention, the laboratories also benefit from eliminating paper processes that are costly to the organization in terms of both time and storage space. In addition, labs benefit from the added control and confidence that all data is being captured, stored, and backed up to be retrieved for an internal or external audit.
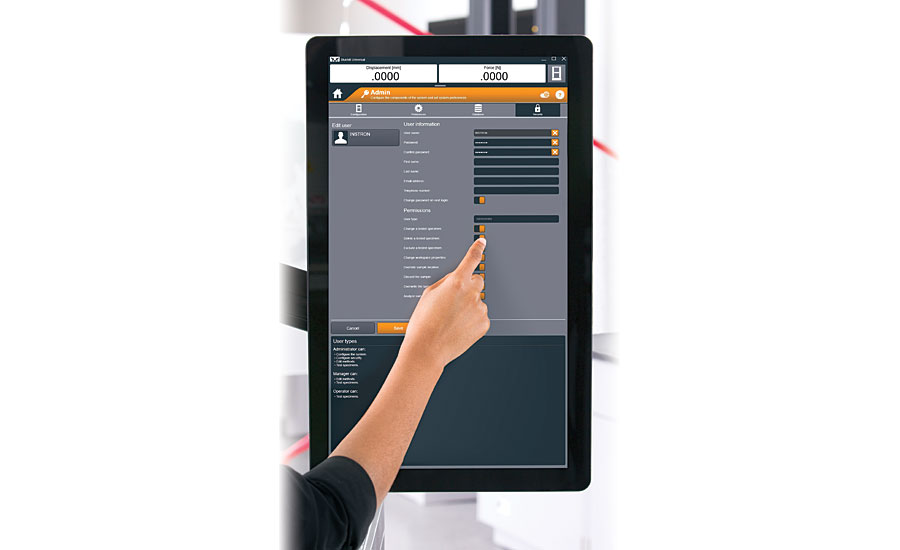
This software features advanced security that allows users to log-in via local Windows accounts, Windows Active Directory, or Bluehill Security. Windows and Active Directory allows the company’s IT department to control password requirements. Bluehill security, pictured above, also allows administrators to set password expiration.
When considering implementing a 21 CFR Part 11 solution in your laboratory and/or moving away from paper records, it is important to remember that your data should be attributable, legible, contemporaneous, original, and accurate. Overall, for medical device and pharmaceutical companies, achieving compliance is best met through collaboration between the end users and the equipment supplier. The end users know how the equipment needs to fit into their quality management systems, and the equipment supplier knows how the system functions and how it can technically meet compliance requirements. If you are a laboratory manager planning a move to electronic records retention and Part 11 compliance, it is important to find equipment suppliers that are willing to work with you in a collaborative process to help define and implement data integrity practices in your lab. Q
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!


